- Атипичный гемолитико-уремический синдром (aГУС)
- Список сокращений
- Термины и определения
- 1. Краткая информация по заболеванию или состоянию (группе заболеваний или состояний)
- 2. Диагностика заболевания или состояния (группы заболеваний или состояний), медицинские показания и противопоказания к применению методов диагностики
- 3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
- 4. Медицинская реабилитация и санаторнокурортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
- 5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
- 6. Организация оказания медицинской помощи
- 7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
- Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение Б. Алгоритмы действий врача
- Список литературы
- Как не пропустить атипичный гемолитикоуремический синдром у пациентов в листе ожидания трансплантации почки
- Генетический профиль пациентов с атипичным гемолитико-уремическим синдромом
Как не пропустить атипичный гемолитикоуремический синдром у пациентов в листе ожидания трансплантации почки
Лекция
Е.И. Прокопенко 1,2
1 ГБУЗ МО Московский областной научно-исследовательский клинический институт им. М.Ф. Владимирского, 129110, Москва, ул. Щепкина, дом 61/2,Российская Федерация 2 ГБУЗ МО
Московский областной научно-исследовательский институт акушерства и гинекологии, 110000 Москва, ул. Покровка, дом 22А, Российская Федерация
Для цитирования: Прокопенко Е.И. Как не пропустить атипичный гемолитико-уремический синдром у пациентов в листе ожидания трансплантации почки. Лекция. Нефрология и диализ. 2024. 26(2):204-215. doi: 10.28996/2618-9801-2024-2-204-215
Ключевые слова: атипичный гемолитико-уремический синдром (аГУС), тромботическая микроангиопатия, трансплантация почки, обследование перед трансплантацией, исходы трансплантации почки, экулизумаб
Резюме
Атипичный гемолитико-уремический синдром (аГУС) – орфанное заболевание, обусловленное дисрегуляцией и гиперактивацией альтернативного пути комплемента, имеющее характерную морфологическую картину поражения сосудов микроциркуляторного русла и триаду (иногда неполную) клинических проявлений: тромбоцитопению, микроангиопатическую гемолитическую анемию и поражение органов-мишеней. Чрезмерная активация комплемента при аГУС вызвана мутациями генов, кодирующих белки системы комплемента, или образованием антител к некоторым из них. Применение комплемент-блокирующей терапии экулизумабом значительно улучшило результаты лечения пациентов с аГУС, в том числе результаты трансплантации почки (ТП) у больных с высоким/средним риском рецидива тромботической микроангиопатии (ТМА) после трансплантации, которым экулизумаб назначается профилактически. Однако некоторые пациенты с аГУС при отсутствии полного симптомокомплекса заболевания достигают ХБП С5 и включаются в листы ожидания ТП без установления правильного диагноза, что может иметь серьезные последствия: возврат аГУС в ренальном трансплантате с быстрой потерей его функции, развитие угрожающих жизни проявлений системной ТМА. Известно, что терапия спасения экулизумабом менее эффективна, чем его профилактическое назначение пациентам группы риска рецидива после ТП. Кроме того, в случае рецидива аГУС в трансплантате время для обследования пациента, как правило, ограничено. В статье приводятся клинические наблюдения, демонстрирующие сложность диагностики «пропущенного» аГУС у пациентов в листе ожидания ТП и после трансплантации. По-видимому, при включении в лист ожидания ТП особую настороженность в отношении аГУС надо проявлять у подростков и пациентов молодого возраста с тяжелой АГ, резистентной к лечению анемией, эпизодами тромбоцитопении, любыми экстраренальными поражениями, пациентов, перенесших типичный ГУС, женщин с ХБП С5 в исходе перенесенной тяжелой преэклампсии/HELLP-синдрома, а также у пациентов, потерявших первый трансплантата по причине ТМА. Таким пациентам должно быть проведено комплексное обследование, включающее повторную оценку анамнеза (в том числе семейного), иммунологическое обследование для исключения других причин ТМА, генетическое исследование системы комплемента, определение антител к фактору H, а также нефробиопсию в отсутствие выраженного уменьшения размеров почек и высокого риска кровотечения.
Введение
Атипичный гемолитико-уремический синдром (аГУС) – орфанное заболевание, вызванное дисрегуляцией и гиперактивацией системы комплемента по альтернативному пути и входящее в группу тромботических микроангиопатий (ТМА) [1]. ТМА является клинико-морфологическим синдромом, характеризующимся своеобразной морфологической картиной поражения сосудов микроциркуляторного русла (в капиллярах – тромбозом и/или эндотелиозом, в артериолах и артериях малого калибра – тромбозом и/или расширением субэндотелиального пространства, мукоидным набуханием интимы с сужением просвета вплоть до окклюзии) и клинической триадой – тромбоцитопенией, микроангиопатической гемолитической анемией (МАГА), поражением органов-мишеней. При ТМА в патологический процесс вовлекаются почки, центральная нервной система (ЦНС), желудочно-кишечный тракт (ЖКТ), сердце, легкие, глаза и др. [2, 3]. В группу ТМА, кроме аГУС, входят тромботическая тромбоцитопеническая пурпура (ТТП), индуцированные инфекциями ТМА (ассоциированный с шигатоксином кишечной палочки ГУС – STEC ГУС (т.н. типичный ГУС), пневмококковый ГУС, ТМА при вирусных инфекциях), а также ТМА, ассоциированные с сопутствующими заболеваниями и состояниями (т.н. вторичные – при системных заболеваниях, осложненной беременности, злокачественных опухолях, применении некоторых лекарств и др.) [4]. Среди взрослых пациентов с ГУС чаще встречается аГУС, у детей, напротив, преобладает STEC ГУС [5].
Чрезмерная активация комплемента при аГУС вызвана мутациями генов, кодирующих белки системы комплемента, или образованием антител к некоторым из них. Развитие аГУС ассоциировано с мутациями с потерей функции генов, кодирующих регуляторные белки – фактор H (CFH), фактор I (CFI), мембранный кофакторный протеин (MCP), а также с генетическими вариантами с усилением функции третьего компонента комплемента (C3) и фактора B (CFB) – двух основных составляющих C3-конвертазы альтернативного пути активации комплемента. Кроме перечисленного выше, патогенетическим фактором аГУС может быть образование аутоантител IgG к CFH, связывающихся с C-терминальным фрагментом CFH и нарушающих его регуляторную функцию. В последние годы выделяют также «комплемент-независимый» аГУС, связанный с мутациями генов, кодирующих не относящиеся непосредственно к системе комплемента белки: плазминоген (PLG), тромбомодулин (THBD), диацилглицеролкиназу ε (DGKE), кобаламин С (cblC), инвертированный формин-2 (INF2) [6]. Однако мутации выявляются не у всех пациентов: при аГУС частота обнаружения генетических вариантов комплемента составляет примерно 60%, при вторичных формах ТМА мутации тоже могут обнаруживаться, хотя и с меньшей частотой [7]. Клинической манифестации аГУС у значительной части пациентов способствует воздействие триггеров (комплемент-активирующих состояний), которыми могут быть инфекции, аутоиммунные заболевания, оперативные вмешательства, некоторые лекарственные препараты, беременность и роды, трансплантация костного мозга или солидных органов [3, 4].
У пациентов с аГУС имеется высокий риск развития рецидивов ТМА. Прогноз для жизни и сохранения почечной функции весьма серьёзен: до начала эры комплемент-блокирующей терапии, когда для лечения аГУС использовалась только плазмотерапия (плазмообмен – ПО и инфузии СЗП), летальность пациентов с аГУС составляла 25%, а частота наступления терминальной ХПН в течение одного года наблюдения – 50% [8]. Особенностью аГУС является и высокая частота рецидивов после трансплантации почки (ТП), создающих угрозу жизни реципиентов и способствующих утрате функции трансплантатов, поэтому всего 10 лет назад ТП не считалась показанной пациентам с терминальной ХПН в исходе аГУС [9]. С появлением экулизумаба (препарата рекомбинантных моноклональных антител против С5-компонента комплемента) ситуация кардинально изменилась в лучшую сторону как в отношении результатов лечения аГУС у пациентов с функционирующими нативными почками, так и в получении возможности безопасно выполнять ТП больным, достигшим терминальной ХПН [10, 11]. Однако продолжает существовать проблема «пропущенного» аГУС у пациентов, нуждающихся в ТП, при этом из всех вариантов ТМА именно аГУС и тяжелые формы STEC ГУС (с неполным восстановлением почечной функции и прогрессированием ХБП после перенесенного ОПП) наиболее часто являются причиной потребности в трансплантационной помощи.
«Тромботическая микроангиопатии неясного генеза» и «пропущенная» тромботическая микроангиопатия у кандидатов на трансплантацию почки
У части пациентов с терминальной ХПН причина ее развития, в том числе ТМА, не устанавливается в ранней стадии заболевания. Несвоевременная диагностики ТМА (в том числе – аГУС) может быть обусловлена разными, как объективными, так и субъективными факторами: нетипичными клиническими симптомами или неполной картиной ТМА, поздним обращением пациентов за медицинской помощью, слабой клинической настороженностью врачей, отсутствием морфологической верификации диагноза у больных с ТМА, поражающей только почки. Напомним, что при локально-почечной ТМА наблюдаются признаки ОПП, а тромбоцитопения, МАГА и экстраренальные проявления отсутствуют [12, 13].
«Пропущенный» аГУС у пациентов с терминальной ХПН, особенно у тех, кому предстоит ТП, может иметь трагические последствия. В то же время, в центры трансплантации направляются больные с установленным «официальным» диагнозом, иногда без достаточного обоснования данного диагноза. Именно поэтому врачи отделений гемодиализа, нефрологи и трансплантологи, которые занимаются отбором пациентов на заместительной почечной терапии (ЗПТ) в лист ожидания ТП, должны проявлять клиническую настороженность в отношении возможной ТМА, в том числе – аГУС.
Перечислим те ситуации, когда можно «пропустить» аГУС у пациентов на диализе и в листе ожидания ТП:
- У взрослых пациентов с «ХПН неясной этиологии» (меньше настороженность в отношении возможности аГУС по сравнению с детьми)
- При отсутствии полного симптомокомплекса МАГА, тромбоцитопении
- При преимущественно почечном поражении без развития полиорганной недостаточности
- При коротком периоде МАГА и спонтанном исчезновении признаков микроангиопатическогоемолиза
- У пациентов с терминальной ХПН неясного генеза, которым не была выполнена биопсия почки, особенно у больных молодого возраста с тяжелой АГ
- При утрате медицинской документации и невозможности полноценного выяснения анамнеза
- При трансформации диагноза (например, аГУС → STEC ГУС) в процессе перехода пациента для наблюдения из одного лечебного учреждения в другое
- У женщин, которые быстро достигли терминальной ХПН после перенесенной тяжелой преэклампсии (ПЭ) с ОПП
Следует особо отметить, что ПЭ, особенно тяжелая, повторная (развивающаяся более чем при одной беременности) является фактором риска развития ХБП в последующей жизни, однако быстрые темпы прогрессирования ХБП все же не характерны для женщин, перенесших это осложнение беременности. А у пациенток, имевших клинически диагностированный (или недиагностированный, протекавший под маской HELLP-синдрома) послеродовый аГУС, даже при отсутствии патогенных мутаций в генах системы комплемента, например, при вероятно патогенных генетических вариантах, или вариантах с неясным значением, существует высокий риск рецидива аГУС после ТП с потребностью в экстренном начале комплемент-блокирующей терапии [14].
аГУС может скрываться не только за диагнозом «ХПН неясной этиологии», но и за диагнозами «Хронический гломерулонефрит», «Быстропрогрессирующий гломерулонефрит», «Системный васкулит», «Гипертонический нефросклероз», «Нефропатия в исходе ОПП», «Типичный (STEC) ГУС».
Мы предлагаем использовать следующий перечень симптомов для оценки пациентов на ЗПТ на предмет возможного аГУС (таблица).
Обнаружение 3 и более признаков, желательно из трех разных пунктов, свидетельствует о том, что подозрение на аГУС оправдано, и требуется дальнейшее обследование. Однако при очевидном, клинически верифицированном эпизоде ТМА в анамнезе или при морфологическом подтверждении ТМА достаточно одного этого признака для констатации высокой вероятности аГУС. Следует понимать, что ценность указанных выше признаков основана только на клиническом опыте автора и не проходила валидацию в силу редкости заболевания и немногочисленности кандидатов с аГУС на трансплантацию.
Таблица Возможные анамнестические, клинико-лабораторные и морфологические признаки у пациентов в листе ожидания трансплантации почки с пропущенным аГУС
| Диагноз, клинические особенности | Терминальная ХПН неясного генеза Типичный ГУС с тяжелым ОПП, неполным восстановлением почечной функции Тяжелая/злокачественная АГ с транзиторным повышением креатинина крови или без него |
| Возраст | Пациенты на ЗПТ молодого возраста* Дети и подростки на ЗПТ |
| Анамнез | Триггер болезни: диарея, респираторные и другие инфекции, вакцинация, лекарства (циклоспорин, такролимус, оральные контрацептивы, противоопухолевые препараты, хинин,
клопидогрел, тиклопидин и др.) Эпизоды анемии и тромбоцитопении в дебюте или на любом этапе болезни У женщин – послеродовое ОПП с неполным восстановлением почечной функции Неуспешная трансплантация почки в анамнезе (особенно первично нефункционирующий трансплантат или потеря трансплантата в первые 6-12 месяцев после операции) Семейный анамнез: наличие родственников с диагностированным аГУС или ТМА, с ХПН неясного генеза или внезапно умерших в молодом возрасте, в том числе – от сердечно-сосудистых катастроф |
| Клиническая картина | Доказанный эпизод (эпизоды) тромбоцитопении и МАГА (анемия, шизоциты в мазке крови, повышение ЛДГ, снижение гаптоглобина, отрицательная проба Кумбса) Наличие любых внепочечных проявлений в дебюте или на любом этапе болезни: поражение ЦНС, глазного дна, кардиомиопатия, сердечная недостаточность, респираторный дистресс-синдром, геморрагический пневмонит, панкреатит, панкреонекроз, поражение ЖКТ, желудочно-кишечное кровотечение, гангрена пальцев Тяжелая, плохо корригируемая АГ с начала болезни Недостаточная реабилитация при адекватной диализной программе: постоянная слабость, утомляемость, невозможность достижения целевого гемоглобина при исключении альтернативных причин резистентности к антианемической терапии Склонность к тромбоцитопении, персистирующее повышение ЛДГ |
| Морфологическая картина | Обнаружение ТМА, в том числе – в сочетании с другими морфологическими диагнозами, в биоптате нативной
почки Обнаружение ТМА, в том числе – в сочетании с другими морфологическими диагнозами, в биоптате трансплантированной почки Обнаружение ТМА в любом другом органе (кроме почки) или в биоптатах кожи |
Подчеркнем, что в сложных случаях отсутствие ТМА при первой нефробиопсии не исключает возможности обнаружения микроангиопатии при повторной биопсии почки, что подтверждает следующее клиническое наблюдение [15].
Авторы из Испании представили 27-летнюю женщину со злокачественной АГ и отягощенным семейным анамнезом: у родной сестры в 14 лет был диагностирован мембранопролиферативный гломерулонефрит с повышением ЛДГ, тяжелой анемией, эпилепсией, ишемическим поражением головного мозга, быстрым прогрессирование ХПН; смерть сестры наступила от легочной инфекции, но данные аутопсии были неопределенными, и основной диагноз не был установлен. Сама пациентка в возрасте одного года перенесла ОПП неясного генеза с анемией и тромбоцитопенией, разрешившееся спонтанно. Однако в 15-летнем возрасте у девочки выявлена АГ с повышением уровня АД до 160/90 мм рт.ст., протеинурия 2 г/сут, умеренное снижение гемоглобина до 115 г/л при нормальном уровне тромбоцитов и сохранной на тот момент почечной функции. Пациентке была выполнена первая нефробиопсия, выявившая фокальный пролиферативный гломерулонефрит с депозитами IgM. Назначена терапия ингибиторами АПФ, но, несмотря на нефропротекцию, отмечен постепенный рост сывороточного креатинина. В возрасте 26 лет внезапно развилась злокачественная АГ (АД 216/120 мм рт.ст.), ретинопатия, начался быстрый рост креатинина, обнаружены тяжелая анемия, тромбоцитопения (тромбоциты 22×109/л), шизоцитоз; активность ADAMTS13 была нормальной. Повторная нефробиопсия подтвердила ренальную ТМА (подострые и хронические сосудистые поражения), которую в то время расценили как следствие злокачественной АГ. Начато лечение ГД, проведено 5 сеансов ПО, после чего микроангиопатический гемолиз был купирован, но сохранялась диализ-зависимая почечная недостаточность и тяжелая АГ, требовавшая одновременного применения семи (!) антигипертензивных препаратов. Три месяца спустя пациентка продемонстрировала снижение интеллекта, кратковременные эпизоды потери сознания и новый эпизод МАГА. При МРТ головного мозга обнаружены очаги гипоксически-ишемического характера, присутствовала также умеренная церебральная атрофия. Генетическое тестирование выявило гомозиготное носительство гаплотипа высокого риска H3 CFH, ассоциированного с низким уровнем фактора H в плазме. Диагноз был пересмотрен: у самой пациентки и ее погибшей сестры (ретроспективно) диагностирован аГУС. Пациентке после отсутствия эффекта от 7 сеансов ПО в виде сохранения тромбоцитопении, МАГА было начато лечение экулизумабом. Гемолиз прекратился после первой же инфузии, после третьего введения препарата отмечено значительное улучшение гематологических показателей и МР-картины головного мозга, однако после 18 мес. лечения пациентка остается зависимой от диализа и ожидает ТП [15].
Описанный испанскими коллегами клинический пример показывает, как трудно порой бывает трактовать клинические симптомы и морфологические данные даже при семейном аГУС. По-видимому, эпизод ОПП в возрасте одного года тоже был обусловлен ТМА, которая впоследствии приняла хроническое течение. Но данные первой нефробиопсии в 15-летнем возрасте скорее затруднили, чем облегчили, постановку диагноза. Вторая биопсия, выполненная уже при развернутой клинической картине МАГА и быстропрогрессирующей почечной недостаточности, выявила признаки острой и хронической ТМА, но причиной ТМА посчитали злокачественную АГ. Лишь в возрасте 27 лет, после начала постоянной ЗПТ, присоединения тяжелого ишемического поражения ЦНС (похожего на поражение головного мозга у сестры) и выполнения генетического исследования удалось установить диагноз аГУС и добиться улучшения после начала комплемент-блокирующей терапии. Понятно, что выполнение ТП у пациентки возможно только при условии применения экулизумаба из-за очень высокого риска рецидива после операции.
Значение своевременной диагностики аГУС у пациентов в листе ожидания трансплантации почки
Быстро установленный диагноз позволяет своевременно начать патогенетическую терапию аГУС. Следует помнить, что именно раннее назначение экулизумаба при аГУС приводит к наиболее полному восстановлению функции нативных почек и других пораженных органов [16]. В чем же опасность отсутствия правильного диагноза у пациентов на ЗПТ без экстраренальных проявлений с необратимой утратой почечной функции в исходе комплемент-опосредованной ТМА? Во-первых, у диализных пациентов могут развиваться тяжелые экстраренальные поражения, связанные с продолжающейся неконтролируемой активацией комплемента и требующие проведения комплемент-блокирующей терапии для спасения жизни, функции ЦНС, зрения. Во-вторых, после ТП риск рецидива аГУС с потерей трансплантата и жизнеугрожающими внепочечными поражениями достаточно высок.
Пациентам с аГУС требуется специальное претрансплантационное обследование, особые подходы к подбору органного донора, к иммуносупрессивной терапии, претрансплантационной вакцинации и медикаментозной профилактике инфекций, плановое принятие решения о необходимости комплементблокирующей терапии. Так, у пациентов с аГУС следует проводить до включения в лист ожидания ТП вакцинацию против менингококковой, пневмококковой и гемофильной инфекции типа b, избегать трансплантации органов от «субоптимальных доноров» и органов с предельно допустимыми сроками холодовой консервации. Проводить генетическое исследование системы комплемента нужно не только у будущего реципиента, но и у потенциального донора (при планировании ТП от живого родственного донора) для исключения риска развития ТМА после изъятия почки [17, 18]. Необходимо применять индукцию иммуносупрессии антитимоцитарными или анти-CD25 антителами, особенно тщательно контролировать концентрации в крови циклоспорина/такролимуса с целью не допустить превышения терапевтических уровней, избегать использования mTOR-ингибиторов (поскольку эта группа иммуносупрессантов, как и ИК, может способствовать развитию ТМА), обязательно проводить профилактику цитомегаловирусной инфекции не менее 6 мес. после операции [18-20]. Части пациентов на основании данных генетического обследования и особенностей семейного анамнеза и клинического течения необходимо при трансплантации профилактическое применение экулизумаба – это пациенты с высоким и средним риском рецидива после ТП [21].
Как правило, у пациентов с аГУС, которым не был установлен диагноз до ТП, при развитии рецидива в трансплантате нет достаточного времени для обследования, при этом возникает проблема дифференциальной диагностики возвратной и de novo ТМА трансплантата, ассоциированной с нефротоксичностью ИК, антитело-опосредованным отторжением, вирусными инфекциями и другими причинами, и далеко не всегда требующей лечения комплемент-блокирующим препаратом [22, 23]. Это, безусловно, не способствует быстрому началу комплемент-блокирующей терапии. Но даже если диагноз аГУС устанавливается после ТП и начинается «терапия спасения» экулизумабом, результаты такого лечения хуже по сравнению с профилактическим применением экулизумаба. Так, Siedlecki A.M. et al. сравнивали потребность в диализе и функцию ренального трансплантата в трех группах пациентов: с диагнозом аГУС, установленным до ТП, и с началом терапии экулизумабом до операции; с диагнозом до ТП, но с началом терапии после операции; с диагнозом, установленным после ТП и с началом терапии после операции. Наиболее высокая потребность в посттрансплантационном диализе и наиболее низкая СКФ трансплантата были именно в последней группе, в который аГУС был диагностирован после трансплантации [24]. В мета-анализе, включившем 18 статей с данным о 380 взрослых пациентах с аГУС после ТП, было показано, что при профилактическом применении экулизумаба частота потерь трансплантатов из-за ТМА составила 5,5%, а при лечении экулизумабом развившихся рецидивов – 22,5%, то есть в 4 раза больше [25].
Наибольшая эффективность применения экулизумаба достигается в том случае, если лечение посттрансплантационного рецидива аГУС начинается в первые 7 дней [26], но при отсутствии диагноза аГУС до операции трудно инициировать комплемент-блокирующую терапию в эти сроки. В ряде случаев происходит очень быстрая потеря трансплантата, как в представленном ниже клиническом наблюдении.
Пациент С., 33 года, поступил в центр трансплантации в феврале 2014 г. для выполнения ТП. В детстве часто болел простудными заболеваниями, имел сниженный аппетит, постоянную слабость, низкий индекс массы тела. Во взрослом возрасте у врача не наблюдался, в 30 лет была случайно обнаружена АГ, антигипертензивные препараты принимал нерегулярно. В 31 года обнаружено повышение креатинина сыворотки до 236 мкмоль/л на фоне повышения АД и незначительной протеинурии. Биопсии почки не было, клинически диагностирован хронический гломерулонефрит, ХПН. Еще через год пациент экстренно доставлен в стационар в тяжелом состоянии с болями в животе, геморрагическим гастритом. При обследовании: гемоглобин 82 г/л, мочевина сыворотки 46 ммоль/л, креатинин 1037 мкмоль/л. Констатирована терминальная ХПН, геморрагический гастрит посчитали проявлением уремии, начато лечение программным ГД. Состояние больного улучшилось, однако сохранялась недостаточно контролируемая АГ и резистентная к лечению анемия, при этом тромбоцитопении, повышения ЛДГ не отмечено. Заподозрен первичный антифосфолипидный синдром (АФС), но маркеры АФС не обнаружены. Пациент был включен в лист ожидания трансплантации почки.
11.02.2014 г. больному была выполнена ТП от донора со смертью мозга, совместимого по группе крови. Предсуществующие антитела у пациента не выявлены, претрансплантационной кросс-матч был отрицательным. Индукция иммуносупрессии проведена базиликсимабом, на операционном столе введен в/в 1 г метилпреднизолона, назначена поддерживающая иммуносупрессия: такролимус, микофеноловая кислота, преднизолон. В первые сутки после операции – гемоглобин 82 г/л, тромбоциты 272×109/л. При наличии кровотока в трансплантате по данным УЗДГ отмечалась олигурия. Через несколько часов после операции (анестезиологическое пособие и операция – без технических проблем и осложнений) появилась неврологическая общемозговая симптоматика: пациент был заторможен, не понимал объяснений лечащего врача, хотя до операции все инструкции выполнял нормально. АД 180/100-110 мм рт.ст. ЦВД 110 мм вод.ст. Натрий сыворотки 136,0 ммоль/л, глюкоза 10 ммоль/л, мочевина 19 ммоль/л. На вторые сутки после операции сохранялась вялость, заторможенность, олигурия, начата ЗПТ (гемодиафильтрация). Отмечено снижение гемоглобина до 66 г/л, тромбоцитов – до 76×109/л, концентрация такролимуса в крови – 14,6 нг/мл. Заподозрена ТМА, ассоциированная с нефротоксичностью ИК, доза такролимуса снижена на 30%, проведена инфузия СЗП. На третьи сутки после ТП: олигурия, АД 190/110 мм рт.ст., гемоглобин 64 г/л, тромбоциты 64×109/л, ЛДГ 1033 ЕД/л. При УЗИ трансплантата отмечалось неравномерное повышение эхогенности коркового слоя, при ДС-ЦК – резкое обеднение кровотока в коре. Диагностирована ТМА, начаты сеансы ПО, назначен низкомолекулярный гепарин, еще больше снижена доза такролимуса. Несмотря на ежедневные сеансы ПО и инфузии СЗП, сохранялись неврологическая симптоматика (МРТ головного мозга – картина отека, мелкие участки кровоизлияний в полушариях большого мозга), тромбоцитопения – 43-79×109/л, повышение ЛДГ до 700 ЕД/л, отсутствовала функции трансплантата. При эхокардиографии выявлено снижении фракции выброса левого желудочка до 40%. На 6-7-е сутки после операции появилась клиника респираторного дистресс-синдрома, начала нарастать дыхательная недостаточность, при этом пациент не был гипергидратирован. ДНК цитомегаловируса, вируса Эпштейна-Барр в крови не обнаружены, прокальцитонин крови не повышен. К сожалению, в 2014 г. еще не было возможности быстро назначить комплемент-блокирующую терапию, и консилиумом принято решение о трансплантатэктомии по витальным показаниям.
На 9-е сутки трансплантат был удален. Морфологическое исследование удаленного трансплантата: картина острой ТМА, признаков отторжения не выявлено. К 14-м суткам после ТП, 5-м суткам после трансплантатэктомии – нормализация уровня тромбоцитов, полный регресс неврологической симптоматики, исчезновение дыхательной недостаточности, восстановление нормальной фракции выброса левого желудочка. Парная почка (от того же донора) после трансплантации пациенту с поликистозом нативных почек продемонстрировала немедленную функцию, течение послеоперационного периода у этого пациента было неосложненным, что позволило нам, с учетом морфологической картины острой ТМА в удаленном трансплантате, исключить наличие донорской патологии. Пациент отказался от дальнейшего обследования и высказал желание оставаться на лечении программным гемодиализом.
После повторного обсуждения данного случая был диагностирован аГУС. К сожалению, пациенту не удалось провести генетическое исследование, но учитывая молниеносное развитие тяжелой системной ТМА после трансплантации почки, отсутствие донорской патологии, отторжения, вирусных инфекций, неэффективность снижения дозы и концентрации ИК в крови, проведения ПО и инфузий СЗП в сочетании с антикоагулянтами, было бы высоко вероятным обнаружение патогенных мутаций в системе комплемента. Сложность диагностики аГУС у данного пациента можно объяснить отсутствием полной клинической картины ТМА до трансплантации, а также отсутствием морфологического диагноза заболевания собственных почек. Нужно отметить, что тяжелая, плохо корригируемая АГ у пациента молодого возраста, анемия, резистентная к лечению, поражение ЖКТ характерны для больных с аГУС, но могут быть вызваны и другими причинами, что в отсутствие тромбоцитопении, явного микроангиопатического гемолиза, мультиорганного поражения затрудняет диагностику.
Может ли типичный гемолитико-уремический синдром оказаться атипичным?
Ранее считалось, что ТП у пациентов, перенесших типичный ГУС, абсолютно безопасна в отношении рецидива ТМА, что представляется логичным с учетом того, что STEC ГУС не рецидивирует [27]. Однако STEC-инфекция может быть триггером ТМА при аГУС, и в этих случаях пациенту может быть ошибочно диагностирован типичный ГУС. Именно такие больные демонстрируют тяжелое течение ТМА с неполным восстановлением почечной функции и быстро достигают тХПН. Однако случаи STEС ГУС тоже могут быть очень тяжелыми, и по клинической картине крайне сложно различить типичный и атипичный ГУС. Дифференциальная диагностика осложняется еще и тем, что в претрансплантационном периоде может не наблюдаться рецидивов ТМА (а STEC ГУС – это болезнь, которая не рецидивирует, в отличие от аГУС), и тогда пациента с ХБП 5 стадии включают в лист ожидания с диагнозом «Типичный ГУС». В этой группе больных первый рецидив ТМА может развиться уже после ТП. По данной причине в настоящее время молекулярно-генетическое исследование системы комплемента перед трансплантацией рекомендуется не только больным с аГУС, но и пациентам с терминальной ХПН в исходе типичного ГУС.
Интересно, что у детей с типичным ГУС патогенные варианты генов комплемента (с частотой встречаемости минорных аллелей в популяции <0,1%) обнаруживались значимо чаще, чем в контрольной группе, и при этом не оказывали влияния на тяжесть течения болезни, в том числе – на поражение ЦНС и потребность в диализе [28]. По-видимому, у детей с такими генетическими вариантами, перенесших типичный ГУС, есть риск рецидива ТМА. Alberti M. et al. описали двух пациентов с диагнозом «STEC ГУС», которые потеряли почечный трансплантат из-за рецидива ТМА. Этим больным генетическое тестирование панели «аГУС» было выполнено уже после утраты трансплантата: у одного пациента выявлена гетерозиготная мутация CFI, а у другой пациентки и её матери, которая стала донором почки, – гетерозиготная мутация MCP [29].
Иногда рецидив ТМА с экстраренальной симптоматикой проходит незамеченным у пациентов на ГД, и диагноз аГУС впервые устанавливается только после ТП. В данном контексте показательно клиническое наблюдение Dowen F. et al., которые описали пациентку 16 лет на программном ГД, у которой тХПН развилась в исходе доказанного типичного ГУС [30]. Через 10 мес. ЗПТ у больной наблюдался судорожный синдром, который в то время был расценен как проявление гипертонической энцефалопатии. Через 18 мес. выполнена ТП, функция трансплантата была отсроченной, а на 10-е сутки после операции у пациентки развилась МАГА, затем судороги, фотофобия, кортикальная слепота; при МРТ головного мозга выявлен PRES-синдром. Заподозрен аГУС, немедленно начато лечение экулизумабом: через 48 ч. после инфузии препарата купировалась неврологические симптомы, через 2 недели наблюдалось восстановление почечной функции и уровня тромбоцитов. Биопсия трансплантата, выполненная на 15-й день после операции, подтвердила наличие ТМА. При генетическом тестировании выявлен редкий гетерозиготный вариант С3 – p.(Ser1619Arg) [30].
Приводим клиническое наблюдение длительного и трудного пути к диагнозу аГУС у пациентки с первоначальным диагнозом STEC ГУС.
Пациентка 1998 г.р. в раннем детском возрасте на фоне острой кишечной инфекции, геморрагического колита перенесла ОПП в сочетании с тяжелой анемией и тромбоцитопенией. Был диагностирован типичный ГУС, проводился перитонеальный диализ, симптоматическая терапия. Полного восстановления функции почек не произошло, в исходе острого эпизода ТМА сформировалась ХБП 3 стадии. В возрасте 8 лет отмечен рост креатинина сыворотки до 650 мкмоль/л, констатирована ХБП 5 стадии, начато лечение постоянным амбулаторным перитонеальным диализом. В феврале 2010 г. в возрасте 12 лет выполнена ТП от посмертного донора, назначена иммуносупрессивная терапия циклоспорином А, микофенолатами и кортикостероидами, функция трансплантата была удовлетворительной. Однако в октябре того же года, через 8 мес. после операции, возникла острая дисфункция трансплантата с повышением креатинина более 200 мкмоль/л, выполнена биопсия пересаженной почки. Морфологически диагностировано острое отторжение (подробное гистологическое описание не сохранилось). Проведено лечение гуморального отторжения: ПО, введение внутривенного иммуноглобулина, ритуксимаба. Была также проведена конверсия с циклоспорина на такролимус. После проведенной терапии уровень креатинина снизился до 140-160 мкмоль/л и сохранялся на таком уровне почти 7 лет.
В 2017 г. в течение 6 мес. креатинин повысился до 300 мкмоль/л. В феврале 2018 г. после переохлаждения отмечено повышение температуры тела до 40°С, сухой кашель, насморк, пациентка самостоятельно принимала НПВС со слабым эффектом. В марте 2018 г. появились боли в подвздошной и поясничной области справа, тошнота, периодически – рвота, сохранялся субфебрилитет, сывороточный креатинин вырос до 900 мкмоль/л. Пациентке начата ЗПТ сначала методом ГД, затем – перитонеального диализа. В апреле 2018 г была выполнена повторная биопсия трансплантированной почки: обнаружено хроническое гуморальное C4d-негативное отторжение, выраженный нефросклероз. По результатам биопсии терапия отторжения признана нецелесообразной, решено продолжить ЗПТ, коррекцию анемии, АД. В апреле-июне 2018 г. самочувствие пациентки оставалось неудовлетворительным, сохранялся субфебрилитет, тошнота, боли в поясничной области. За это время трижды девушка находилась на стационарном лечении, в начале июля выписана по семейным обстоятельствам в связи со смертью матери.
В июле 2018 г. – повышение температуры тела до 38,5°С, увеличение трансплантата в размерах, расширение чашечно-лоханочной системы. Ситуация в целом трактовалась как обострение хронического пиелонефрита нефункционирующего трансплантата, поэтому была выполнена трансплантатэктомия, начата антибактериальная терапия. Через 7 дней после выписка из стационара, где было выполнено удаление трансплантата, появились выраженная слабость, рвота, затем присоединился повторный жидкий стул. АД 140/80 мм рт.ст., гемоглобин 85 г/л, тромбоциты 92×109/л, лейкоциты 9,2×109/л; шизоцитоз +++, ЛДГ 700-474 ЕД/л; протромбиновый индекс 55%, МНО 1,41, фибриноген 2,7 г/л. При КТ головного мозга выявлены внутричерепные гематомы: слева в лобной области размером 18 мм, справа – единичные до 4 мм. На рентгенограмме грудной клетки легкие полнокровны, корни расширены. Диагностированы ОНМК (геморрагический инсульт) и ТМА, назначены антигипертензивная терапия, низкомолекулярный гепарин, цитофлавин, эритропоэтин; начат перитонеальный диализ, плазмотерапия не проводилась. Постепенно состояние пациентки улучшилось, ТМА регрессировала. Пациентке был продолжен перитонеальный диализ, поставлен вопрос о повторной ТП.
При обследовании перед повторной трансплантацией для уточнения причины ТМА исследованы активность ADAMTS13 в плазме, антикардиолипиновые антитела, антитела к β2-гликопротеину I, волчаночный антикоагулянт. Данных за ТТП и АФС не получено. Был уточнен семейный анамнез: мать пациентки страдала с подросткового возраста неконтролируемой тяжелой АГ, перенесла повторные ОНМК и умерла в возрасте 44 лет (июнь 2018) при клинической картине ТМА. У самой пациентки при генетическом исследовании на хромосоме 1 обнаружена микроделеция 1q31.3(196743721-196801783)x1 протяжённостью 58063 пары нуклеотидов CFHR1,CFHR3 в гетерозиготном состоянии. Данная мутация может быть ассоциирована с развитием аГУС. Диагноз был пересмотрен, констатирован аГУС, семейная форма заболевания. Рекомендовано не использовать для трансплантации почку от родственника без генетического обследования донора, при ТП начать профилактическое применением экулизумаба.
В данном наблюдении с момента первого эпизода ТМА до установления диагноза аГУС прошло 20 лет. Примечательно, что пациентка не продемонстрировала картину ТМА в раннем посттрансплантационном периоде, а затем на фоне гуморального отторжения почечного трансплантата, чего можно было бы ожидать, поскольку сама операция ТП и антитело-опосредованное отторжение являются мощными триггерами ТМА. Первый рецидив аГУС произошел после трансплантатэктомии по поводу пиелонефрита нефункционирующего трансплантата. При ведении пациентки поначалу не был учтен семейный анамнез – высокая АГ с подросткового возраста и повторные ОНМК у матери.
Обследование пациентов с подозрением на тромботическую микроангиопатию перед трансплантацией почки
Пациенты с подозрением на ТМА нативных почек или с морфологически подтвержденной ТМА неясного генеза перед включением в лист ожидания ТП должны пройти дополнительное обследования с целью подтверждения/исключения ТМА, а также установления причины ТМА. Как уже было отмечено выше, крайне важно диагностировать аГУС до трансплантации с учетом высокой вероятности тяжелых осложнений и быстрой потери трансплантата при использовании обычных протоколов ведения реципиентов ренального трансплантата. Предлагаем следующий алгоритм обследования пациентов с подозрением на аГУС и другие формы ТМА перед включением в лист ожидания ТП (рисунок).
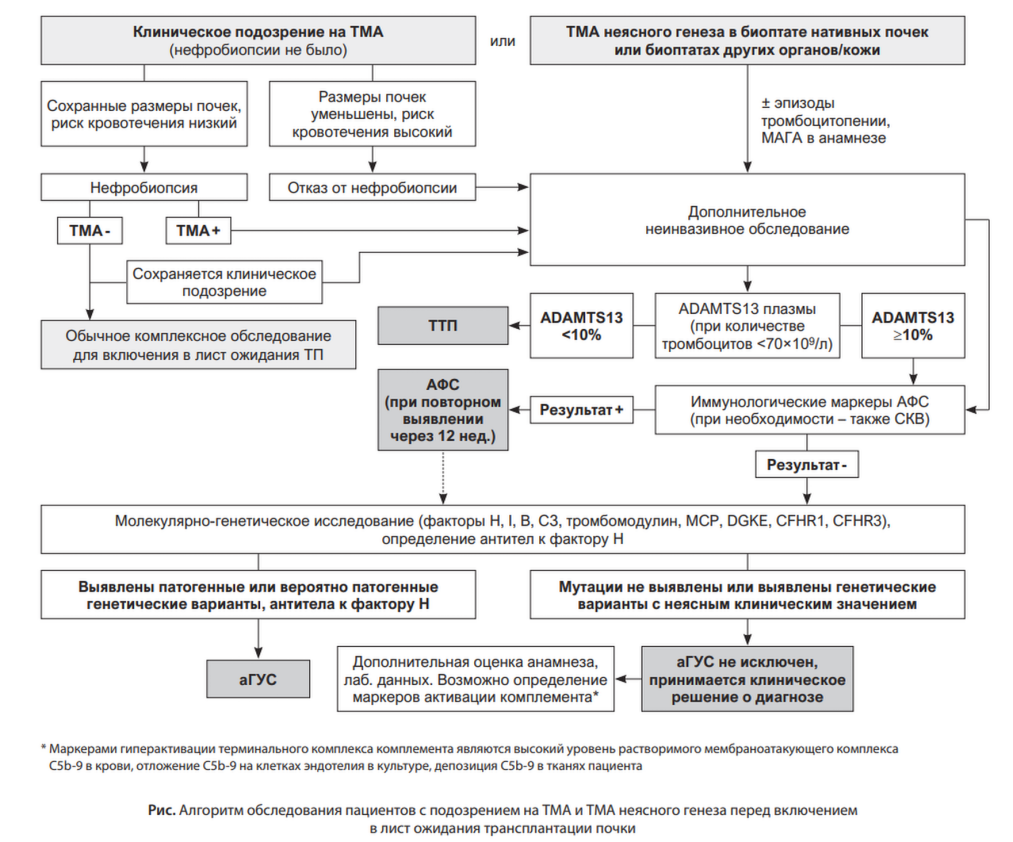
При типичной клинической картине аГУС для подтверждения диагноза не требуется биопсия почки. Но в диагностически сложных ситуациях полезно получить морфологическое подтверждение ТМА, и проведение ГД само по себе не является препятствием к нефробиопсии. Однако, как известно, ХПН (особенно с существенным уменьшением размеров почек), тромбоцитопения, сама ренальная ТМА и целый ряд других факторов повышают риск образования больших постпункционных гематом и даже продолжительного кровотечения из места пункции, хотя частота серьезных геморрагических осложнений после нефробиопсии по данным мета-анализа невысока – гемотрансфузии требовались в 1,6% случаев, а вмешательства для остановки кровотечения – в 0,3% [31, 32]. По указанным причинам вопрос о проведении биопсии почки при подозрении на ТМА у пациентов на ГД решается индивидуально, с тщательной оценкой соотношения польза/риск инвазивного диагностического вмешательства и наличия возможности купирования вероятных осложнений в данном лечебном учреждении. Как правило, если пациент находится на ЗПТ уже долго, более 3-4 мес., от выполнения биопсии отказываются. Кроме того, еще раз напомним, что биопсия выявляет только наличие ТМА, но не верифицирует ее причину. При типичном и атипичном ГУС, АФС, склеродермическом почечном кризе, лекарственной ТМА морфологическая картина почечной ткани будет практически одинаковой [33].
Если по результатам биопсии ренальная ТМА исключена (не учитываются биопсии с недостаточным количеством материала, неопределенным заключением морфолога), перед включением в лист ожидания проводится обычное обследование. Следует подчеркнуть, что при сохранении серьезного клинического подозрения дополнительное неинвазивное обследование тоже проводится даже при отрицательных результатах морфологического исследования, как и в тех случаях, когда ТМА обнаружена. Это обследование должно включать определение активности ADAMTS13 и наличия ингибитора ADAMTS13 в плазме у пациентов с тромбоцитопенией – уровнем тромбоцитов ниже 70×109/л, исследование крови на антикардиолипиновые антитела, антитела к β2-гликопротеину I, волчаночный антикоагулянт (при положительном результате для подтверждения диагноза АФС обследование необходимо повторить через 12 недель) [34]. Поскольку значительная часть случаев АФС наблюдается у пациентов с СКВ, целесообразно провести также исследование иммунологических маркеров волчанки. Конечно, следует исключить и системную склеродермию, но все-таки пациенты со склеродермией обычно имеют клинический диагноз. В отдельных, достаточно редких случаях, у пациента может быть выявлена не одна болезнь, например, волчаночный нефрит и аГУС или IgA-нефропатия и аГУС [35-37].
При исключении ТТП, СКВ, АФС (а в некоторых случаях и при выявлении волчаночных и антифосфолипидных антител, поскольку пациенты с СКВ и АФС могут иметь вторичный аГУС с наличием мутаций) необходимо провести молекулярно-генетическое исследование панели «аГУС» и, крайне желательно, определение антител к фактору H. В случае выявления патогенных или вероятно патогенных генетических вариантов комплемента при наличии ТМА пациенту устанавливается диагноз аГУС, и проводится соответствующая профилактика рецидива заболевания при трансплантации. Сложность заключается в том, что отсутствие генетических вариантов системы комплемента или выявление мутаций с неясным значением не исключает аГУС [38]. В подобных ситуациях следует еще раз провести подробную оценку личного и семейного анамнеза пациента, клинической картины, обратиться за помощью в экспертный центр. Если клинико-анамнестически устанавливается ТМА и при этом исключаются другие причины микроангиопатии, пациенту ставится диагноз аГУС даже при отрицательных результатах генетического тестирования.
Дополнительными аргументами в пользу аГУС могут быть биомаркеры активации комплемента, определение которых пока не вошло в широкую клиническую практику, но может быть очень полезным для подтверждения комплемент-опосредованного характера ТМА: это высокий уровень растворимого мембраноатакующего комплекса (C5b-9) в крови, депозиция C5b-9 в культуре клеток эндотелия и в тканях пациента (положительное окрашивание биоптата при иммуногистохимическом исследовании на C5b-9 и C4d) [33, 39].
Заключение
В последнее десятилетие количество публикаций, посвященных аГУС и ТМА значительно увеличилось, что способствовало улучшению диагностики аГУС. У многих врачей появился собственный опыт ведения пациентов с ТМА. Тем не менее, остаются проблемы диагностики аГУС, особенно при отсутствии полного симптомокомплекса, развития локально-почечной ТМА. Некоторые пациенты с аГУС достигают терминальной ХПН и включаются в листы ожидания трансплантационных центров без установления правильного диагноза. Выполнение ТП таким больным с применением обычных протоколов ведения чревато серьезными последствиями – рецидивом ТМА в почечном трансплантате, быстрой потерей функции трансплантированной почки, развитием системной ТМА с угрожающими жизни органными поражениями. Нередко только после потери первого трансплантата впервые возникает подозрение на аГУС, при этом в условиях дефицита донорских органов во всех странах мира, возможности сенсибилизации пациента (появления анти-HLA антител) после первой ТП, возникновения негативного отношения пациента к ТП после неудачи первой операции, выполнение повторной трансплантации может стать серьезной проблемой.
По описанным выше причинам перед нефрологами, специалистами по диализу, трансплантологами, которые направляют пациентов на ТП или участвуют в работе комиссий по включению пациентов в лист ожидания ТП, стоит важная задача – не пропустить аГУС у пациентов с неясными причинами терминальной ХПН. Особое внимание, по-видимому, необходимо уделять подросткам и пациентам молодого возраста с тяжелой АГ, резистентной к лечению анемией, эпизодами тромбоцитопении, любыми экстраренальными поражениями, пациентам, перенесшим типичный ГУС, женщинам, у которых почечная недостаточность развилась после перенесенной тяжелой преэклампсии/HELLP-синдрома, а также пациентам, потерявшим свой первый ренальный трансплантат из-за ТМА. В диагностике аГУС и определении дальнейшей тактики у кандидатов на ТП с неясным диагнозом большую роль играют не только высокотехнологичные методы обследования, такие как генетическое исследование системы комплемента, иммунологические методы обследования, морфологическая диагностика, но и тщательная оценка анамнеза, в том числе семейного (при необходимости – запрос медицинских документов из других лечебных учреждений), анализ особенностей течения болезни, исключение других причин ХБП 5 стадии. Применение мультидисциплинарного подхода с привлечением экспертов различных специальностей, коллегиальное обсуждение сложных клинических случаев может помочь ускорить диагностику и улучшить как результаты трансплантации почки, так и прогноз заболевания в целом у пациентов с пропущенным аГУС.
Список литературы
1. Thompson G.L., Kavanagh D. Diagnosis and treatment of thrombotic microangiopathy. Int J Lab Hematol. 2022. 44(Suppl 1):101-113. doi: 10.1111/ijlh.13954
2. Brocklebank V., Wood K.M., Kavanagh D. Thrombotic microangiopathy and the kidney. Clin J Am Soc Nephrol. 2018. 13(2):300-317. doi: 10.2215/CJN.00620117
3. Gallan A.J., Chang A. A new paradigm for renal thrombotic microangiopathy. Semin Diagn Pathol. 2020. 37(3):121-126. doi: 10.1053/j.semdp.2020.01.002
4. McFarlane P.A., Bitzan M., Broome C. et al. Making the correct diagnosis in thrombotic microangiopathy: a narrative review. Can J Kidney Health Dis. 2021. 22;8: 20543581211008707. doi: 10.1177/20543581211008707
5. Palma L.M., Vaisbich-Guimarães M.H., Sridharan M. et al. Thrombotic microangiopathy in children. Pediatr Nephrol. 2022. 1-14. doi: 10.1007/s00467-021-05370-8
6. Åkesson A., Martin M., Blom A.M. et al. Clinical characterization and identifi cation of rare genetic variants in atypical hemolytic uremic syndrome: A Swedish retrospective observational study. Ther Apher Dial. 2021. 25(6):988-1000. doi: 10.1111/1744- 9987.13634
7. Palma L.M., Sridharan M., Sethi S. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. 2021. 6(1):11-23. DOI: 10.1016/j.ekir.2020.10.009
8. Noris M., Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009. 361(17):1676-1687. doi: 10.1056/ NEJMra0902814
9. Le Quintrec M., Zuber J., Moulin B. et al. Complement genes strongly predict recurrence and graft outcome in adult renal transplant recipients with atypical hemolytic and uremic syndrome. Am J Transplant. 2013. 13:663–675. doi: 10.1111/ ajt.12077
10. Greenbaum L.A., Fila M., Ardissino G. et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016. 89(3):701-711. doi: 10.1016/j.kint.2015.11.026
11. Tang Z.C., Hui H., Shi C., Chen X. New fi ndings in preventing recurrence and improving renal function in AHUS patients after renal transplantation treated with eculizumab: a systemic review and meta-analyses. Ren Fail. 2023. 45(1):2231264. doi: 10.1080/0886022X.2023.2231264
12. Hanna R.M., Henriksen K., Kalantar-Zadeh K. et al. Thrombotic microangiopathy syndromes-common ground and distinct frontiers. Adv Chronic Kidney Dis. 2022. 29(2):149-160. e1. doi: 10.1053/j.ackd.2021.11.006
13. Saba E.S., Cambron J.C., Go R.S. et al. Clinical associations, treatment, and outcomes of renal-limited thrombotic microangiopathy. Blood. 2018. 132 (Suppl_1): 4978. doi: 10.1182/ blood-2018-99-117723
14. Garlo K., Dressel D., Savic M., Vella J. Successful eculizumab treatment of recurrent postpartum atypical hemolytic uremic syndrome after kidney transplantation. Clin Nephrol Case Stud. 2015. 3:8-13. doi: 10.5414/CNCS108491
15. Ávila A., Vizcaíno B., Molina P. et al. Remission of aHUS neurological damage with eculizumab. Clin Kidney J. 2015. 8(2): 232–236. doi: 10.1093/ckj/sfu144
16. Brocklebank V., Walsh P.R., Smith-Jackson K. et al. Atypical hemolytic uremic syndrome in the era of terminal complement inhibition: an observational cohort study. Blood. 2023. 142(16):1371-1386. doi: 10.1182/blood.2022018833
17. Claes K.J., Massart A., Collard L. et al. Belgian consensus statement on the diagnosis and management of patients with atypical hemolytic uremic syndrome. Acta Clin Belg. 2018. 73(1):80-89. doi: 10.1080/17843286.2017.1345185
18. Tseng M.H., Lin S.H., Tsai J.D. et al. Atypical hemolytic uremic syndrome: Consensus of diagnosis and treatment in Taiwan. J Formos Med Assoc. 2023. 122(5):366-375. doi: 10.1016/j. jfma.2022.10.006
19. Прокопенко Е.И. Применение эверолимуса у de novo реципиентов почечного трансплантата. Вестник трансплантологии и искусственных органов. 2010. 12(2): 74- 81. Prokopenko E.I. Everolimus in de novo renal transplant recipients. Russian Journal of Transplantology and Artifi cial Organs. 2010. 12(2):74-81. (in Russ.).
20. Прокопенко Е.И., Щербакова Е.О., Ватазин А.В. и др. Результаты профилактики цитомегаловирусной инфекции валганцикловиром у пациентов с трансплантированной почкой. Клиническая нефрология. 2013. 5:37-41. Prokopenko E.I., Shcherbakova E.O., Vatazin A.V. et al. Valganciclovir in cytomegalovirus prophylaxis in renal transplant recipients. Clinical Nephrology. 2013. 5:37-41. (in Russ.).
21. Козловская Н.Л., Добронравов В.А., Боброва Л.А. и др. Клинические рекомендации по ведению взрослых пациентов с атипичным гемолитико-уремическим синдромом. Нефрология и диализ. 2023. 25(4):465-492. doi: 10.28996/2618- 9801-2023-4-465-492 Kozlovskaya N.L., Dobronravov V.A., Bobrova L.A. et al. Clinical guidelines for the management of adult patients with atypical hemolytic-uremic syndrome. Nephrologу and Dialуsis. 2023. 25(4):465-492. doi: 10.28996/2618-9801-2023-4-465-492. (in Russ.).
22. Teixeira C.M., Tedesco Silva Junior H., Moura L.A.R. et al. Clinical and pathological features of thrombotic microangiopathy infl uencing long-term kidney transplant outcomes. PLoS One. 2020. 15(1):e0227445. doi: 10.1371/journal.pone.0227445
23. Imanifard Z., Liguori L., Remuzzi G. et al. TMA in kidney transplantation. Transplantation. 2023. 107(11):2329-2340. doi: 10.1097/TP.0000000000004585
24. Siedlecki A.M., Isbel N., Vande Walle J. et al. Eculizumab use for kidney transplantation in patients with a diagnosis of atypical hemolytic uremic syndrome. Kidney Int Rep. 2018. 4(3):434- 446. doi: 10.1016/j.ekir.2018.11.010
25. Gonzalez Suarez M.L., Thongprayoon C., Mao M.A. et al. Outcomes of kidney transplant patients with atypical hemolytic uremic syndrome treated with eculizumab: a systematic review and meta-analysis. J Clin Med. 2019. 8(7):919. doi: 10.3390/ jcm8070919
26. Walle J.V., Delmas Y., Ardissino G. et al. Improved renal recovery in patients with atypical hemolytic uremic syndrome following rapid initiation of eculizumab treatment. J Nephrol. 2017. 30(1):127-134. doi: 10.1007/s40620-016-0288-3
27. Ferraris J.R., Ramirez J.A., Ruiz S. et al. Shiga toxinassociated hemolytic uremic syndrome: absence of recurrence after renal transplantation. Pediatr Nephrol. 2002. 17(10):809-14. doi: 10.1007/s00467-002-0936-9
28. Frémeaux-Bacchi V., Sellier-Leclerc A.L., Vieira-Martins P. et al. Complement gene variants and shiga toxin-producing Escherichia coli-associated hemolytic uremic syndrome: retrospective genetic and clinical study. Clin J Am Soc Nephrol. 2019. 14(3):364-377. doi: 10.2215/CJN.05830518
29. Alberti M., Valoti E., Piras R. et al. Two patients with history of STEC-HUS, posttransplant recurrence and complement gene mutations. Am J Transplant. 2013. 13(8):2201-2206. doi: 10.1111/ajt.12297
30. Dowen F., Wood K., Brown A.L. et al. Rare genetic variants in Shiga toxin-associated haemolytic uraemic syndrome: genetic analysis prior to transplantation is essential. Clin Kidney J. 2017. 10(4):490-493. doi: 10.1093/ckj/sfx030
31. Poggio E.D., McClelland R.L., Blank K.N. et al. Systematic review and meta-analysis of native kidney biopsy complications. Clin J Am Soc Nephrol. 2020. 15(11):1595-1602. doi: 10.2215/CJN.04710420
32. Halimi J.-M. Complications after native kidney biopsy: defi nitive data. Curr Opin Nephrol Hypertens. 2021. 30(6):555- 558. doi: 10.1097/MNH.0000000000000736
33. Palma L.M.P., Sridharan M., Sethi S. Complement in secondary thrombotic microangiopathy. Kidney Int Rep. 2021. 6(1):11-23. doi: 10.1016/j.ekir.2020.10.009
34. Miyakis S., Lockshin M.D., Atsumi T. et al. International consensus statement on an update of the classifi cation criteria for defi nite antiphospholipid syndrome (APS). J Thromb Haemost. 2006. 4(2):295-306. doi: 10.1111/j.1538-7836.2006.01753.x
35. Kim M.J., Lee H., Kim Y.H. et al. Eculizumab therapy on a patient with co-existent lupus nephritis and C3 mutationrelated atypical haemolytic uremic syndrome: a case report. BMC Nephrol. 2021. 22(1):86. doi: 10.1186/s12882-021-02293-2
36. Alhamoud I., Freiberg S.A. Successful discontinuation of eculizumab in a pediatric patient with atypical hemolytic uremic syndrome and underlying systematic lupus erythematosus. Cureus. 2022. 14(5):e25117. doi: 10.7759/cureus.25117
37. Manenti L., Rossi G.M., Pisani I. et al. IgA nephropathy and atypical hemolytic uremic syndrome: a case series and a literature review. J Nephrol. 2022. 35(4):1091-1100. doi: 10.1007/ s40620-021-01189-6
38. Fremeaux-Bacchi V., Fakhouri F., Garnier A. et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013. 8(4):554-562. doi: 10.2215/CJN.04760512
39. Blasco M., Guillén E., Quintana L.F. et al. Thrombotic microangiopathies assessment: mind the complement. Clin Kidney J. 2020. 14(4):1055-1066. doi: 10.1093/ckj/sfaa195