- Атипичный гемолитико-уремический синдром (aГУС)
- Список сокращений
- Термины и определения
- 1. Краткая информация по заболеванию или состоянию (группе заболеваний или состояний)
- 2. Диагностика заболевания или состояния (группы заболеваний или состояний), медицинские показания и противопоказания к применению методов диагностики
- 3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
- 4. Медицинская реабилитация и санаторнокурортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
- 5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
- 6. Организация оказания медицинской помощи
- 7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
- Приложение А1. Состав рабочей группы по разработке и пересмотру клинических рекомендаций
- Приложение А2. Методология разработки клинических рекомендаций
- Приложение Б. Алгоритмы действий врача
- Список литературы
- Как не пропустить атипичный гемолитикоуремический синдром у пациентов в листе ожидания трансплантации почки
- Генетический профиль пациентов с атипичным гемолитико-уремическим синдромом
Генетический профиль пациентов с атипичным гемолитико-уремическим синдромом
Е.С. Иванова1, Е.С. Столяревич1, 2, О.Н. Котенко1, В.Е. Виноградов1, Л.Ю. Артюхина1, Н.Ф. Фролова1, П.А. Шаталов3, В.В. Ильинский4
1 Московский городской научно-практический центр нефрологии и патологии трансплантированной почки «ГБУЗ ГКБ № 52» ДЗМ, Москва, Россия 2 Кафедра нефрологии ФПДО ФГБУ ФГОУ «Московский государственный медико-стоматологический университет им. А.И. Евдокимова», Москва, Россия 3 ФГБУ «НМИЦ радиологии» Минздрава России, г. Обнинск, Россия 4 ООО «Генотек», Москва, Россия
Резюме
Целью настоящего исследования стал анализ спектра генетических мутаций у пациентов с аГУС.
Материал и методы. Ретроспективное исследование по результатам генетического исследования 44 пациентов с аГУС, наблюдавшихся на базе Научно-практического центра нефрологии и патологии трансплантированной почки ГКБ №52 с 2014 по 2021 гг.
Результаты. У 68% пациентов с аГУС были выявлены мутации в системе комплемента. Среди характерных для аГУС мутаций чаще всего встречались мутации генов CFHR1, CFHR3 –13 пациентов (30%). Реже наблюдались мутации в генах С3 – 11 пациентов (26%), CFH – 8 пациентов (19%), CD46 – 2 пациента (5%). У 5 из 7 пациенток с акушерским аГУС были обнаружены характерные для аГУС генетические мутации (CFHR1, CFHR3, CFH, CFI, C3, CD46, THBD). Среди 11 пациентов с аГУС в собственных почках без заместительной почечной терапии у 6 были обнаружены патогенные мутации, которые в большинстве случаев (4 пациента) были представлены мутациями CFHR1, CFHR3. В группе пациентов с аГУС и терминальной ХПН патогенные мутации обнаружены у 8 из 10 пациентов, в группе аГУС и АТП – у 11 из 16 пациентов. В обеих группах чаще выявлялись мутации С3, CFH и CFHR1, CFHR3, а среди поражения органов-мишеней преобладало поражение сердца.
Заключение. Полученные данные предоставляют информацию о генетических причинах болезни и корреляциях генотипа и фенотипа, которые могут прогнозировать прогрессирование заболевания, ответ на терапию и риск рецидива после трансплантации. Это позволяет осуществлять индивидуальный подход к ведению пациентов и лечению, основанному на экспертной интерпретации генетических профилей, что требует проведения генетического скрининга у каждого пациента.
Ключевые слова: атипичный гемолитико-уремический синдром, тромботическая микроангиопатия, комплемент, генетическое исследование.
Сведения об авторах:
Иванова Екатерина Сергеевна: к.м.н., врач-нефролог нефрологического отделения №1 ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: katerineiv@mail.ru; ORCID: https://orcid.org/0000-0001-7407-5695.
Столяревич Екатерина Сергеевна: д.м.н., проф. кафедры нефрологии ФПДО ФГБОУ ВО «МГМСУ им. А.И. Евдокимова» МЗ РФ, врач-патологоанатом отделения патологической анатомии ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: Stolyarevich@yandex.ru; ORCID: https://orcid.org/0000-0002-0402-8348.
Котенко Олег Николаевич: к.м.н., главный внештатный специалист нефролог ДЗМ, руководитель Научно-практического центра нефрологии и патологии трансплантированной почки ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: olkotenko@yandex.ru; ORCID: https://orcid.org/0000-0001-8264-7374.
Артюхина Людмила Юрьевна: к.м.н., заведующая нефрологическим отделением №1 ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: arlyu-1404@yandex.ru, ORCID: https://orcid.org/0000-0003-3353-1636.
Виноградов Владимир Евгеньевич: заведующий консультативно-диагностическим нефрологическим отделением ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: vino-gradoff@yandex.ru, ORCID: https://orcid.org/0000-0002-0184-346Х.
Фролова Надия Фяатовна: к.м.н., заместитель главного врача по нефрологической помощи ГБУЗ «ГКБ № 52» ДЗМ; 123182, г. Москва, ул. Пехотная, д.3.; e-mail: nadiya.frolova@yandex.ru; ORCID: http://orcid.org/0000-0002-6086-5220.
Шаталов Петр Алексеевич: к.б.н., руководитель молекулярно-генетической службы ФГБУ «НМИЦ радиологии» Минздрава России 249036, Калужская область, г.Обнинск, ул.Королева, д.4. ORCID: https://orcid.org/0000-0001-5374-8547
Ильинский Валерий Владимирович: генеральный директор ООО «Генотек», 105120, Москва, Наставнический пер., 17 стр.1, подъезд 14, ORCID: https://orcid.org/0000-0003-4377-2759
Атипичный гемолитико-уремический синдром (аГУС) представляет собой тяжелое системное заболевание, характеризующееся тромбоцитопенией, гемолитической анемией и острым повреждением почек, которое индуцируется гиперактивацией комплемента по альтернативному пути [1, 2]. Это очень редкое заболевание с частотой примерно 0,5 случаев на миллион человек в год. До появления экулизумаба (гуманизированного моноклонального антитела против С5, которое блокирует терминальный путь каскада комплемента) оно имело очень плохой прогноз [3, 4]. Являясь типичным заболеванием, опосредованным комплементом, аГУС развивается у людей с предрасполагающими генетическими аномалиями в генах комплемента после воздействия множества провоцирующих/причинных событий, включая инфекции, лекарства, злокачественные новообразования, трансплантацию и беременность [5].
Генетические исследования у пациентов с клиническим диагнозом аГУС выявляют мутации в генах, связанных с альтернативным путем активации системы комплемента, почти в половине всех случаев [6, 7]. Список широко известных генов при аГУС включает CFH (причастен примерно у 25% пациентов), CD46 или MCP (около 10%), C3 (около 6%), CFI (около 6%), CFB (около 2%), THBD (около 2%), и не связанный с комплементом ген диацилглицеролкиназы эпсилон – DGKE (приблизительно 3%) [5]. Аутоантитела к фактору H обнаруживаются в 5–13% случаев и связаны с отсутствием обеих копий CFHR1 [8]. Если обнаруживается генетическая вариация, ее часто считают предрасполагающим фактором, а не непосредственной причиной аГУС. Это различие отражает высокую вариабельность пенетрантности болезни, за исключением патогенных вариантов DGKE, которые подчиняются аутосомно-рецессивному типу наследования [9].
При выявлении мутации генов у пациента с аГУС крайне важно определить их клиническую значимость, так как это имеет отношение к длительности антикомплементарной терапии. Выявляемые варианты мутаций в генах, связанных с альтернативным путем активации комплемента, не во всех случаях имеют отношение к патогенезу аГУС. В ряде публикаций было показано, что ультраредкие, но доброкачественные варианты не являются патогенными [10]. Например, Marinozzi и соавторы экспериментально продемонстрировали, что 9 из 15 зарегистрированных мутаций гена CFB не были связаны с патогенезом аГУС [11]. Также были идентифицированы новые варианты мутаций CFH, которые могут быть не связаны с аГУС [12]. Эти отчеты подтверждают ценность функциональных исследований для оценки влияния разных генетических вариантов, которые являются достаточно трудоемкими и сложными, что делает функциональную оценку непрактичной в каждом случае.
Цель исследования
Изучение спектра генетических мутаций у пациентов с аГУС.
Материалы и методы
На базе Научно-практического центра нефрологии и патологии трансплантированной почки ГКБ №52 проведено ретроспективное исследование, в которое были включены 44 пациента (28 мужчин и 16 женщин) с аГУС, наблюдавшихся в клинике с 2014 по 2021 гг. Средний возраст пациентов составил 29,6 ± 11,1 лет.
Все пациенты с установленным диагнозом аГУС условно были разделены на 4 группы. 1 группу представляли пациентки с акушерским аГУС (7 пациенток). Во 2 группу входили пациенты с аГУС, диагностированным в собственных почках (11 пациентов) и которые не нуждались в заместительной почечной терапии (ЗПТ) на момент последнего наблюдения. 3 группа была представлена пациентами с аГУС и терминальной хронической почечной недостаточностью (ТХПН) на лечении программным диализом (10 пациентов). Некоторым пациентам этой группы диагноз аГУС был окончательно верифицирован после проведения генетического исследования, так как клинически течение заболевания почек, приведшее к ТХПН, заставляло подозревать аГУС. В связи с планируемой трансплантацией почки у данной группы пациентов генетический анализ стал ключевым для определения тактики их ведения в посттрансплантационном периоде. 4 группа была образована пациентами с аГУС после аллотрансплантации трупной почки (АТП) (16 пациентов).
Пациентам всех групп проведены физикальный осмотр, лабораторно-инструментальное обследование (клинический и биохимический анализы крови, общий анализ мочи и суточный анализ мочи, ЭКГ, ЭХОКГ, УЗИ почек и органов брюшной полости). Ряду пациентов по показаниям дополнительно проведены МСКТ и МРТ головного мозга, ЭГДС и ФКС. 25 пациентам (57%) была выполнена пункционная биопсия почки с проведением световой микроскопии и иммунофлюоресцентного исследования на замороженных срезах. У всех 25 пациентов по результатам биопсии почки выявлены признаки тромботической микроангиопатии (ТМА).
По клинической картине и результатам лабораторно-инструментальных методов исследования пациентам проводилась оценка наличия поражения органов-мишеней помимо почек в рамках аГУС (сердце, головной мозг, кишечник и другие органы).
Всем пациентам был выполнен генетический анализ крови на панель аГУС. ДНК выделялась с помощью стандартных методик наборами реагентов Qiagen на системах автоматического выделения ДНК QIAcube и Freedom EVO. Качественные и количественные показатели образца ДНК измерялись с помощью гель-электрофореза нуклеиновых кислот и с интеркалирующим красителем на флуориметре Qubit. ДНК ферментативно разрезали на фрагменты определенной длины, к которым добавлялись специальные последовательности, называемые адаптерами. Библиотеки ДНК готовили с использованием роботизированной станции Tecan Freedom EVO и амплификатора T100. Далее оценивали количество и качество созданной библиотеки ДНК. Качественный анализ выполняли методом капиллярного гель-электрофореза на приборах Bioanalyzer 2100 (Agilent) или LabChip GX Touch (Perkin Elmer). Количественный анализ проводили методом ПЦР в реальном времени на амплификаторе StepOnePlus. Необходимые для последующего анализа последовательности генома обогащали с помощью биотинилированных РНК-зондов и анализировали на приборах HiSeq 2500 и MiSeq по протоколу производителя. После контроля качества полученных данных (точность прочтения, глубина и полнота прочтения), проводился поиск генетических вариантов. В качестве референсного генома использовалась сборка GRCh38.p13. Каждому обнаруженному варианту присваивалась одна из пяти категорий патогенности или доброкачественности согласно рекомендациями ACMG. Врач-генетик составлял список генетических вариантов, связанных с клиническими симптомами пациента, на основании данных клинической картины или направительного диагноза пациента.
Выделение ДНК, приготовление и секвенирование ДНК-библиотек
Выделение ДНК проводили с помощью набора “QIAmp DNA mini kit” (Qiagen). Молекулярную массу геномной ДНК подтверждали, используя гель-электрофорез, при этом следили за отсутствием деградации ДНК и загрязнения РНК. Концентрацию выделенной ДНК определяли используя Qubit 3.0 (Life Technologies). ДНК-библиотеки готовили с использованием набора NEBNext® Ultra™ DNA Library Prep Kit for Illumina (New England Biolabs), используя адаптеры последовательности для секвенирования на приборах Illumina, согласно стандартному протоколу, предложенному производителем. Двукратное индексирование выполняли с помощью ПЦР с NEBNext Multiplex Oligos for Illumina® (Dual Index Primers Set 1). Качество и концентрацию библиотек определяли с помощью Bioanalyzer 2100 (Agilent Technologies). Для обогащения фракции геномной ДНК использовали набор The SureSelect XT2 Target Enrichment System (Agilent Technologies). ДНК-библиотеки секвенировали на геномном анализаторе HiSeq2500 (Illumina) с использованием парных чтений с длиной чтения 100 нуклеотидов.
Биоинформатический анализ
Нуклеотиды с 3’-конца, имеющие качество прочтения ниже 10, обрезались с помощью программы cutadapt. Выравнивание на референсный геном hg19 (GRCh37.p13) было выполнено с помощью bwa. Дедупликация ридов выполнена с помощью samtools rmdup. Контроль качества чтений осуществляли с помощью программ FastQC и Genotek quality analysis solution. Распознавание коротких вариантов (изменений, мутаций) осуществляли GATK HaplotypeCaller согласно установленному протоколу GATK DNA-seq. Для рассмотрения выбирались лишь те варианты, покрытие в которых составляло ≥ 12. Влияние каждого изменения оценивали с помощью snpEff, оценки патогенности и консервативности, данные для которых извлекали из баз данных dbNSFP, Clinvar, OMIM и HGMD, а также с использованием утилит SIFT и polyphen2 для предсказания возможной патогенности мутации. Информация о частоте мутаций бралась из 1000Genomes project, ExAC и Genotek frequency data. Аннотация мутаций и их патогенность предсказывалась согласно Стандартам и Руководству, разработанным ACMG, AMP и CAP для интерпретации мутаций, полученных с помощью секвенирования. Количество копий мутаций и цитогенетические перестановки осуществляли с помощью CNVkit.
Обнаруженные у пациентов мутации были условно разделены на 3 группы: мутации, характерные для аГУС (CFH, CFI, C3, CD46 (MCP), THBD, DGKE, CHFR1/CHFR3), мутации тромбофилий (MTHFR, ITGA2, ITGB3, F5, F7, F13A1, FGB) и мутации ADAMTS13.
Статистические расчеты (описательная статистика) выполнены с помощью программы Excel и статистической программы SPSS Statistics 23.
Результаты
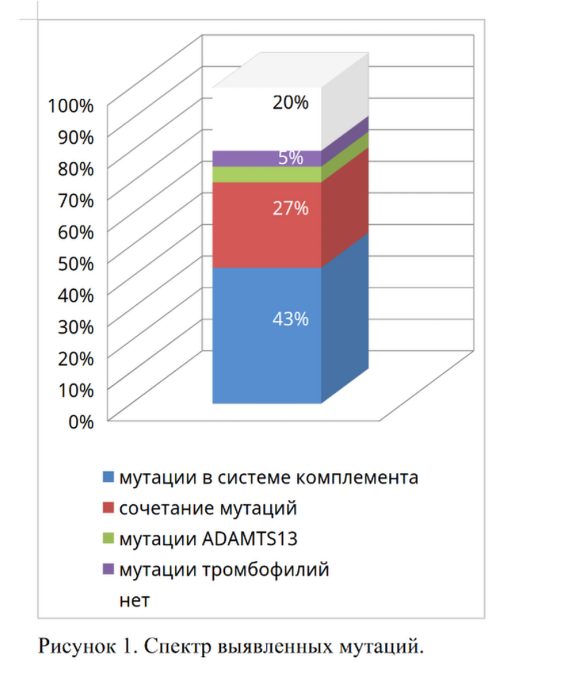
Среди пациентов с аГУС мутации в системе комплемента были обнаружены у 30 из 44 пациентов (68%). Мутации тромбофилий выявлены у 14 пациентов (32%). При этом у 11 пациентов (25% от общего числа) наблюдалось сочетание мутаций в системе комплемента с мутациями тромбофилий. Изолированные мутации в системе комплемента обнаружены у 19 пациентов (43%), изолированные мутации тромбофилии – у 2 пациентов (5%). У 6 пациентов (14%) выявлены мутации ADAMTS13 (у 2 пациентов изолировано, у 3 – в сочетании с мутациями системы комплемента и тромбофилий, у 1 – в сочетании с мутациями тромбофилий). У 9 пациентов (20%) мутации не обнаружены (Рисунок 1).
Среди характерных для аГУС мутаций чаще всего встречались мутации генов CFHR1, CFHR3 – у 13 пациентов (30%). При этом чаще наблюдалось именно сочетание CFHR1, CFHR3. Изолированные мутации в этой группе наблюдались значительно реже: CFHR1 – 2 пациента (5%), CFHR3 – 2 пациента (5%), CFHR4 – 1 пациент (2%) и CFHR5 – 1 пациент (2%). Вторыми по частоте у наших пациентов были мутации в генах С3 – 11 пациентов (26%). У 8 пациентов (19%) были обнаружены мутации в генах CFH. У 2 пациентов (5%) выявлены мутации CD46 (MCP). Остальные мутации встречались значительно реже (Рисунок 2а).
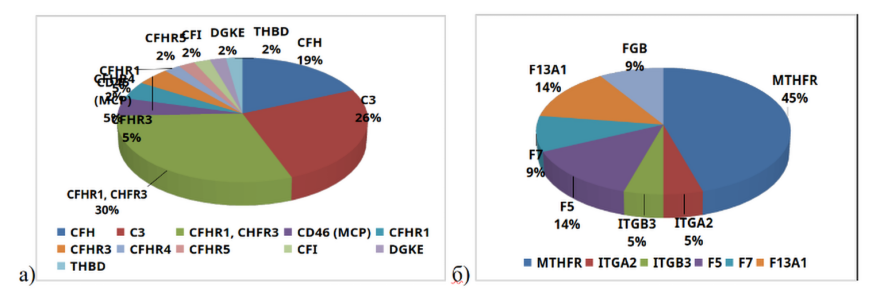
Рисунок 2. Мутации у пациентов с аГУС: а) характерные для аГУС мутации, б) мутации генов, ассоциированных с тромбофлией.
Помимо известных патогенных для аГУС мутаций был проведен анализ других мутаций (чаще всего ассоциированных с тромбофилией), обнаруженных у наших пациентов с аГУС (Рисунок 2б). В большинстве случаев это были мутации MTHFR – 10 пациентов (45%). У 14% пациентов были мутации F5 и F13А1, реже встречались мутации FGB и F7 (по 9%), ITGB3 (5%) и ITGA2 (4%).
Также был проведен анализ различных мутаций и поражений органов-мишеней по выделенным группам пациентов (Рисунок 3).
У 5 из 7 пациенток с акушерским аГУС были обнаружены характерные для аГУС генетические мутации (CFH, CFI, C3, CD46, THBD), при этом у трех из них эти мутации сочетались с тромбофилическими мутациями и мутациями ADAMTS13 (Рисунок 3а). Чаще всего у пациенток с акушерским аГУС наблюдалось изолированное поражение почек, но было по 1 пациентке с поражением сердца, кишечника или ЦНС. Биопсия почки выполнялась только одной пациентке и подтвердила наличие ТМА. 6 из 7 пациенток данной группы начали своевременное лечение экулизумабом. При этом у 4 пациенток функция почек полностью восстановилась, 2 из них имели мутации в генах CHFR1, CHFR3, CD46 и С3, у 2 других патогенных мутаций не было. У 1 пациентки почечная функция восстановилась до уровня начальной ХПН, у нее были мутации CHFR1, CHFR3 и THBD. 1 пациентка с мутациями CHFR1, CHFR3 осталась на лечении программным гемодиализом. Единственной пациентке данной группы, которой не было своевременно начато лечение экулизумабом, диагноз аГУС был установлен еще в период, когда лечение экулизумабом в нашей стране было не доступно. Ей была выполнена АТП, однако через 2 года в связи с рецидивом аГУС в трансплантате она вернулась на лечение программным гемодиализом. У нее были обнаружены мутации генов CFH и CFI. При повторной трансплантации назначена терапия экулизумабом, на фоне чего почечный трансплантат удовлетворительно функционирует в течение 6,5 лет.
Среди 11 пациентов с аГУС в собственных почках без ЗПТ у шестерых были обнаружены патогенные мутации, которые в большинстве случаев (у 4 пациентов) были представлены мутациями CFHR1, CFHR3 (Рисунок 3б). В данной группе нередко наблюдалось сочетание поражения почек с поражением кишечника и ЦНС (по 4 пациента), сердца (3 пациента). У 4-х пациентов выполнялась биопсия почки, подтвердившая ТМА. Все пациенты данной группы получают лечение экулизумабом. На фоне лечения экулизумабом 3 из 11 пациентов полностью восстановили функцию почек. Среди этих 3-х пациентов у двух были мутации CFHR1, CFHR3, у одного патогенных мутаций не было обнаружено. Средний уровень креатинина крови остальных 8 пациентов составил 211 ± 140 мкмоль/л.
В группе пациентов с аГУС и ТХПН у 8 из 10 пациентов были обнаружены патогенные мутации (Рисунок 3в). В большинстве случаев это были мутации С3, CFH и CFHR1, CFHR3. Нередко наблюдалось сочетание патогенных мутаций с мутациями ADAMTS13 и тромбофилий, среди которых чаще всего встречались мутации MTHFR и F5. У 7 пациентов представленной группы наблюдалось поражение сердца в рамках аГУС, реже выявлялись поражения кишечника и ЦНС. У 8 из 10 пациентов была выполнена биопсия почки, подтвердившая ТМА. 5 из 10 пациентов получают терапию экулизумабом, что обусловлено наличием у них тяжелого поражения других органов-мишеней в рамках аГУС (сердце, ЦНС). Остальным пациентам планируется лечение экулизумабом после выполнения АТП.
Патогенные мутации в группе аГУС и АТП выявлены у 11 из 16 пациентов (Рисунок 3г). При этом чаще всего были обнаружены мутации С3, CFHR1, CFHR3 и CFH, как и в группе пациентов с аГУС и ТХПН. Нередко наблюдалось сочетание с мутациями MTHFR. В данной группе среди поражения органов-мишеней преобладало поражение сердца (выявлено у 7 пациентов), как и в группе с аГУС и ТХПН. Диагноз ТМА подтвержден по биопсии собственных почек у 3 пациентов, у 4 пациентов – по биопсии трансплантированной почки. Все 16 пациентов получают лечение экулизумабом. Среди этих 16 пациентов 10 пациентам диагноз был поставлен еще до выполнения АТП и терапию экулизумабом они начали получать сразу после операции. У этих пациентов выявлялись мутации CFH, CFI, C3, CD46 (MCP), CFHR1, CFHR3, CFHR5. К настоящему времени у всех этих пациентов трансплантаты функционируют и средний уровень креатинина крови в данной группе пациентов составляет 125 ± 29 мкмоль/л. Остальным 6 пациентам диагноз аГУС был поставлен уже после АТП. Трое из этих пациентов, у которых определялись мутации CFHR1, CFHR3, CFH, C3, вернулись на лечение программным диализом, несмотря на проводимую антикомплементарную терапию. У оставшихся 3-х пациентов с мутациями CFHR1, CFHR3, С3 средний уровень креатинина крови составляет 208 ± 105 мкмоль/л.
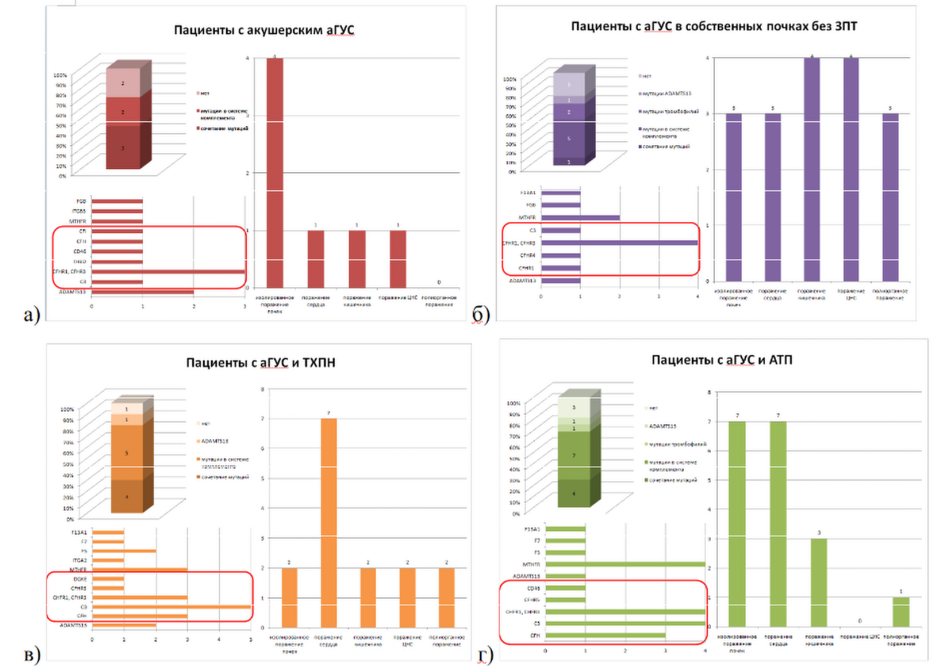
Рисунок 3. Спектр генетических мутаций и поражений органов-мишеней в группах пациентов с аГУС: а) пациенты с акушерским аГУС, б) пациенты с аГУС в собственных почках без ЗПТ, в) пациенты с аГУС и ТХПН, г) пациенты с аГУС и АТП. Красной рамкой выделены патогенные мутации.
Обсуждение
Выявление патогенного генетического варианта у пациента с аГУС подтверждает диагноз и позволяет с точностью установить причину заболевания, облегчая ведение пациента, эффективное лечение и генетическое консультирование.
Скрининг редких вариантов генов комплемента обычно включает секвенирование не менее пяти генов комплемента – CFH, C3, CFI, CFB и MCP, а также выявление гибридных генов CFHR1-CFHR3, вызывающие неаллельные гомологичные рекомбинации [13]. На сегодняшний день у пациентов с аГУС идентифицировано более 500 вариантов этих пяти генов комплемента [14]. Эти гены кодируют белки, которые участвуют в регуляции как на клеточной поверхности, так и в жидкой фазе альтернативного пути комплемента, за исключением MCP, который участвует только в регуляции на клеточной поверхности альтернативного пути комплемента [15]. Результаты исследований, в ходе которых был проведен скрининг редких вариантов гена комплемента у более чем 3000 пациентов во всем мире, показали, что аГУС является заболеванием, опосредованным комплементом. Примечательно, что во всех опубликованных сериях примерно половина обнаруженных вариантов была локализована в гене CFH [14].
Так в итальянском исследовании Noris с соавторами, включавшем 273 пациента с аГУС (73% пациентов из Европы, 16% из Америки, 2% из Африки, 1% их Азии, 8% с Ближнего Востока) в период с 1996 до 2007 гг. мутации в системе комплемента были обнаружены примерно у 70% пациентов. При этом чаще всего наблюдались мутации CFH – 23%, реже мутации MCP – 7%, THBD – 5%, C3 – 4% и CFI – 4%. Пациенты с мутациями CFH или THBD имели самое раннее начало и самую высокую смертность. Мутации MCP были связаны с лучшим прогнозом. Плазмотерапия индуцировала ремиссию в 55–80% случаев у пациентов с мутациями CFH, C3, THBD или аутоантителами, в то время как пациенты с мутациями CFI плохо реагировали на лечение [9].
Во французском исследовании среди 214 пациентов с аГУС, наблюдавшихся в период с 2000 по 2008 гг., у 60,2% были выявлены мутации в системе комплемента. Среди них чаще всего также встречались мутации CFH – 28%, реже мутации MCP – 9%, CFI – 8%, С3 – 8% и CFB – 2% [16].
В американском исследовании 144 пациентов с аГУС имелось схожее распределение по мутациям: CFH – 27%, CFI – 8%, MCP – 5%, CFB – 4%, THBD – 3% и C3 – 2% [17].
Несколько иное распределение частоты мутаций получилось в японском исследовании Fujisawa с соавторами. В исследование было включено 118 японских пациентов в период с 1998 по 2016 гг. У них наиболее частыми генетическими аномалиями были мутации C3 (31%), а частота мутаций CFH была относительно низкой (10%) по сравнению с западными странами. В этой когорте преобладал вариант С3 p.I1157T (23%), что было связано с благоприятными исходами, несмотря на частые рецидивы. В общей сложности 72% пациентов получали плазмотерапию, а 42% лечили экулизумабом. Прогноз у японских пациентов с аГУС был относительно благоприятным, с общей смертностью 5,4% и почечной смертностью 15% [18].
В нашем исследовании мутации в системе комплемента были обнаружены у 68% пациентов с аГУС, что соотносится с международными данными. По спектру выявленных мутаций у наших пациентов чаще наблюдались мутации С3 (26%), что больше соответствует данным японской группы пациентов. С другой стороны, частота обнаружения мутаций CFH (19%) скорее ближе к результатам западных исследователей, хоть и представлена несколько более низким процентом.
В группе пациенток с акушерским аГУС мутации в системе комплемента наблюдались у 71% пациенток. В работе Fakhouri с соавторами был проведен анализ 21 пациентки с акушерским аГУС: мутации в системе комплемента были обнаружены у 86%, при этом чаще, чем в общей популяции, выявлялись мутации CFH (45%) [19]. В нашей группе пациенток с акушерским аГУС мутации CFH, CFI, C3, CD46, THBD наблюдались с одинаковой частотой – 14%, что, вероятно, обусловлено небольшим количеством пациенток.
У пациентов с аГУС с поражением собственных почек без ЗПТ не было обнаружено серьезных мутаций CFH, CFI, которые, как известно, значимо ухудшают прогноз и часто приводят к ТХПН. Однако у этих пациентов нередко наблюдались экстраренальные проявления с поражением кишечника, ЦНС и сердца.
Интересно, что пациенты групп ТХПН и АТП по своему спектру генетических мутаций и экрстраренальным проявлениям очень схожи. Это обусловлено тем, что по своей сути пациенты из группы ТХПН постепенно переходят в группу пациентов с АТП и, наоборот, – все пациенты с АТП еще недавно были пациентами с ТХПН.
При аГУС сообщается об изменениях генов CFHR1, CFHR3 и фактора H в сочетании с интактными генами CFHR2, CFHR4 и CFHR5. А изменения в каждом из пяти генов CFHR в контексте интактного гена фактора H описаны при С3-гломерулопатии [20]. Белки CFHR с 1 по 5 структурно сходны с фактором H и кодируют области, которые расположены рядом с геном CFH на хромосоме 1. Гены CFHR, считающиеся псевдогенами, имеют высокую степень гомологии последовательностей с CFH и, следовательно, восприимчивы к генетическим перестройкам, приводящим к образованию гибридов CFH-CFHR. При этом их пенетрантность составляет около 50% как в семейных, так и в спорадических случаях, и для развития заболевания у людей с данными мутациями требуется серьезный триггер [21].
Гомозиготная делеция CFHR3-CFHR1 наблюдается примерно у 15% преимущественно молодых пациентов с аГУС. У этих пациентов часто обнаруживаются аутоантитела к фактору Н. Точные механизмы, посредством которых при аГУС гомозиготный дефицит CFHR1 и CFHR3 индуцирует выработку антител, неясны. Один из механизмов заключается в том, что FHR3 связывается с фрагментом C3d и блокирует адъювантный эффект C3d при активации В-клеток [22].
В индийской статье Kandari с соавторами [23] были проанализированы результаты генетического исследования 17 пациенток с акушерским аГУС. У 11 пациенток были обнаружены гетерозиготные делеции CFHR1 и CFHR3, у 4 пациенток – гомозиготные делеции CFHR1 и CFHR3. И лишь у 2 пациенток мутаций не было.
Согласно нашим данным, у пациенток с акушерским аГУС мутации CFHR1, CFHR3 также являлись наиболее частыми генетическими аномалиями в данной группе – у 3 из 7 пациенток. Как известно, беременность и роды являются очень серьезным триггером, что, по всей видимости, обуславливает высокую частоту встречаемости данной аномалии у пациенток с акушерским аГУС. Кроме того, мутации CFHR1 и CFHR3 нередко наблюдались и в остальных группах пациентов с аГУС. К сожалению, по техническим причинам не всем этим пациентам был выполнен анализ крови на антитела к фактору Н. Среди 4-х обследованных пациентов с подобными генетическими аномалиями у двух были обнаружены антитела к фактору Н.
Вклад тромбофилических мутаций в развитии аГУС не установлен. Однако, как следует из представленных данных у пациентов с аГУС помимо мутаций в системе комплемента нередко можно обнаружить тромбофилические мутации. В литературе описаны мутации при аГУС, которые не связаны с системой комплемента. Примерами являются мутации в генах диацилглицеролкиназы эпсилон (DGKE), инвертированного формина-2 (INF2) и плазминогена (PLG). У наших пациентов встречались мутации DGKE, а вот мутации INF2 и PLG не были обнаружены, что обусловлено все-таки достаточно редкой встречаемостью этих аномалий.
Внепочечные проявления аГУС наблюдаются примерно у 20% пациентов [24]. При этом могут поражаться многие системы органов, в том числе периферическая и центральная нервная система (8-48%), желудочно-кишечный тракт (8-10%), сердечно-сосудистая система (3-10%), кожа, легкие (около 21%), глаза (4%). Некоторые из этих проявлений возникают в острой фазе аГУС, другие могут быть проявлением отдаленных последствий бесконтрольной активации комплемента.
Согласно нашим данным у пациенток с акушерским аГУС в большинстве случаев наблюдалось изолированное поражение почек. У пациентов с аГУС в собственных почках без ЗПТ среди экстраренальных проявлений чаще встречались поражения кишечника и ЦНС (по 36%). А в группах ТХПН и АТП экстраренальные проявление были схожими и чаще представлены поражением сердца (70% и 44% соответственно). Поражение сердца у этих пациентов может быть как прямым следствием активации комплемента и ТМА, так и усугубляться на фоне тяжелой артериальной гипертензии и уремии [5], которые наблюдаются у пациентов с ТХПН и через которые проходят все пациенты с АТП.
В группе пациентов после АТП вне зависимости от выявленных мутаций исходы по большей части были связаны со своевременностью начала терапии экулизумабом. Так, у пациентов, которым лечение экулизумабом было начато сразу после АТП в качестве профилактической терапии, почечные трансплантаты сохранили свою функцию, несмотря на наличие таких мутаций, как CFH, CFI, C3, которые, как известно, определяют частые рецидивы аГУС в трансплантате при отсутствии антикомплементарной терапии [25, 26]. Среди пациентов, которым лечение экулизумабом было начато при развитии рецидива аГУС в почечном трансплантате, половина из них потеряли свои трансплантаты и вернулась на лечение диализом. При этом генетические мутации у этих пациентов были схожи (в том числе мутации CFH, C3), что подтверждает необходимость своевременной диагностики аГУС (по возможности – до выполнения АТП) и раннего начала комплемент-блокирующей терапии у этой группы пациентов.
Заключение
Исследование сотен пациентов с аГУС предоставило информацию о генетических причинах болезни и корреляциях генотипа и фенотипа, которые могут прогнозировать прогрессирование заболевания, ответ на терапию и риск рецидива после трансплантации. Это позволяет осуществлять индивидуальный подход к ведению пациентов и лечению, основанному на экспертной интерпретации генетических профилей, что требует проведения генетического скрининга у каждого пациента. Задержки в получении результатов генетических или молекулярно-диагностических исследований не должны препятствовать постановке клинического диагноза и началу лечения, так как ранняя антикомплементарная терапия имеет решающее значение для сохранения функции почек и предотвращения необратимых последствий.
Никто из авторов не имеет конфликтов интересов
Вклад авторов:
Концепция и дизайн исследования – Иванова Е.С., Столяревич Е.С., Котенко О.Н.
Сбор и обработка материала – Иванова Е.С., Виноградов В.Е., Артюхина Л.Ю., Фролова Н.Ф., Шаталов П.А., Ильинский В.В.
Статистическая обработка данных – Иванова Е.С.
Написание текста – Иванова Е.С.
Редактирование – Столяревич Е.С.
Список литературы:
1. Nester CM, Barbour T, de Cordoba SR, Dragon-Durey MA, Fremeaux-Bacchi V, Goodship TH, et al. .: Atypical aHUS: State of the art. Mol Immunol 67: 31–42, 2015 [PubMed: 25843230]
2. Bu F, Zhang Y, Wang K, Borsa NG, Jones MB, Taylor AO, Takanami E, Meyer NC, Frees K, Thomas CP, Nester C, Smith RJH. Genetic Analysis of 400 Patients Refines Understanding and Implicates a New Gene in Atypical Hemolytic Uremic Syndrome. J Am Soc Nephrol. 2018 Dec;29(12):2809-2819. doi: 10.1681/ASN.2018070759.
3. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. .: Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 368: 2169–2181, 2013 [PubMed: 23738544]
4. Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A: A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open 3: e003573, 2013 [PMCID: PMC3822313] [PubMed: 24189082]
5. Goodship TH, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. .: Conference Participants : Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) controversies conference. Kidney Int 91: 539–551, 2017 [PubMed: 27989322]
6. Kavanagh D, Goodship TH, Richards A: Atypical hemolytic uremic syndrome. Semin Nephrol 33: 508–530, 2013 [PMCID: PMC3863953] [PubMed: 24161037]
7. Jokiranta TS: HUS and atypical HUS. Blood 129: 2847–2856, 2017 [PMCID: PMC5445567] [PubMed: 28416508]
8. Durey MA, Sinha A, Togarsimalemath SK, Bagga A: Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol 12: 563–578, 2016 [PubMed: 27452363]
9. Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859. doi:10.2215/CJN.02210310
10. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T,et al. .: Exome Aggregation Consortium : Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291, 2016 [PMCID: PMC5018207] [PubMed: 27535533]
11. Marinozzi MC, Vergoz L, Rybkine T, Ngo S, Bettoni S, Pashov A, et al. .: Complement factor B mutations in atypical hemolytic uremic syndrome-disease-relevant or benign? J Am Soc Nephrol 25: 2053–2065, 2014 [PMCID: PMC4147975] [PubMed: 24652797]
12. Merinero HM, García SP, García-Fernández J, Arjona E, Tortajada A, Rodríguez de Córdoba S: Complete functional characterization of disease-associated genetic variants in the complement factor H gene. Kidney Int 93: 470–481, 2018 [PubMed: 28941939]
13. Fakhouri, F., Zuber, J., Fremeaux-Bacchi, V. & Loirat, C. Haemolytic uraemic syndrome. Lancet 217, 681–696 (2017).
14. Osborne, A. J. et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 glomerulopathy. J. Immunol. 200, 2464–2478. (2018).
15. Merle, N. S., Church, S. E., Fremeaux-Bacchi, V. & Roumenina, L. T. Complement system Part I — molecular mechanisms of activation and regulation. Front. Immunol. 6, 262 (2015).
16. Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8(4):554-562. doi:10.2215/CJN.04760512.
17. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010 Jun;31(6):E1445-60. doi: 10.1002/humu.21256.
18. Fujisawa M, Kato H, Yoshida Y, et al. Clinical characteristics and genetic backgrounds of Japanese patients with atypical hemolytic uremic syndrome [published correction appears in Clin Exp Nephrol. 2019 Apr 23;:]. Clin Exp Nephrol. 2018;22(5):1088-1099. doi:10.1007/s10157-018-1549-3
19. Fakhouri F, Roumenina L, Provot F, et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutations. J Am Soc Nephrol. 2010;21(5):859-867. doi:10.1681/ASN.2009070706.
20. Zipfel PF, Wiech T, Stea ED, Skerka C. CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J Am Soc Nephrol. 2020;31(2):241-256. doi:10.1681/ASN.2019050515
21. Nozawa, A., Ozeki, M., Hori, T., Kawamoto, N., Hirayama, M., Azuma, E., & Fukao, T. (2018). A Heterozygous CFHR3-CFHR1 Gene Deletion in a Pediatric Patient With Transplant-associated Thrombotic Microangiopathy Who was Treated With Eculizumab. Journal of pediatric hematology/oncology, 40(8), e544–e546. https://doi.org/10.1097/MPH.0000000000000986
22. Zipfel, Peter F et al. “CFHR Gene Variations Provide Insights in the Pathogenesis of the Kidney Diseases Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy.” Journal of the American Society of Nephrology : JASN vol. 31,2 (2020): 241-256. doi:10.1681/ASN.2019050515.
23. Kandari S, Chakurkar V, Gaikwad S, Agarwal M, Phadke N, Lobo V. High prevalence of CFHR deletions in Indian women with pregnancy-associated hemolytic uremic syndrome. Nephrology (Carlton). 2022;27(3):231-237. doi:10.1111/nep.14004
24. Formeck C, Swiatecka-Urban A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr Nephrol. 2019;34(8):1337-1348. doi:10.1007/s00467-018-4039-7.
25. Noris M, Remuzzi G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr Opin Nephrol Hypertens. 2013;22:704–712.
26. Noris M, Bresin E, Mele C, Remuzzi G. Genetic Atypical Hemolytic-Uremic Syndrome. 2007 Nov 16 [updated 2021 Sep 23]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Mirzaa GM, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2022. PMID: 20301541.